 Las proteínas no son solo nutrientes presentes en los alimentos. Consisten en largas y complejas moléculas, compuestas de aminoácidos, que llevan a cabo la mayoría de las funciones en el interior de las células, como la replicación del material genético, la obtención de energía o la señalización de todas las rutas indispensables para su funcionamiento. Una de las características más importantes de las proteínas es que su función depende de cómo se pliegan: sus propiedades físico-químicas las hacen adquirir una estructura tridimensional, sin la que no pueden funcionar.
Las proteínas no son solo nutrientes presentes en los alimentos. Consisten en largas y complejas moléculas, compuestas de aminoácidos, que llevan a cabo la mayoría de las funciones en el interior de las células, como la replicación del material genético, la obtención de energía o la señalización de todas las rutas indispensables para su funcionamiento. Una de las características más importantes de las proteínas es que su función depende de cómo se pliegan: sus propiedades físico-químicas las hacen adquirir una estructura tridimensional, sin la que no pueden funcionar.
Por eso, desde hace 50 años uno de los retos más importantes de la Biología ha sido precisamente averiguar cómo se pliegan las proteínas, lo que tiene infinitas aplicaciones en la investigación básica, en procesos industriales y en el campo del desarrollo de fármacos: por ejemplo, conocer cómo se pliega la proteína S del coronavirus es crucial para diseñar vacunas u otros medicamentos.
Hoy se conocen alrededor de 180 millones de proteínas, pero solo se ha podido averiguar cómo se pliegan 170.000 de ellas. Esto se ha logrado por medio de técnicas experimentales, como la cristalografía de rayos X, que las «congela» y cristaliza, para resolver su estructura con fotones, o la resonancia magnética. Pero, como este trabajo es muy complicado y algunas proteínas «se resisten», también se trabaja en modelos y en aproximaciones de bioinformática para predecir el plegamiento de las proteínas a partir de su secuencia de aminoácidos. Pero hay tantas posibilidades y dificultades en cada proteína que hasta ahora no se ha logrado un gran desarrollo en el campo.
Todo esto parece haber cambiado. Esta semana, DeepMind, filial de Google, que ha creado sistemas de Inteligencia Artificial (IA) capaces de aprender y vencer en el ajedrez, el go o en videojuegos de disparos, ha desarrollado otra IA, de nombre AlphaFold, capaz de predecir la estructura tridimensional de las proteínas. En concreto, ha logrado alcanzar una precisión del 92% en esta tarea, en un encuentro bianual destinado a poner a prueba a modelos bioinformáticos, y de nombre CASP, de «Critical Assessment of protein Structure Prediction». Sus resultados se han anunciado esta semana, pero todavía no se han publicado en una revista científica revisada por pares.
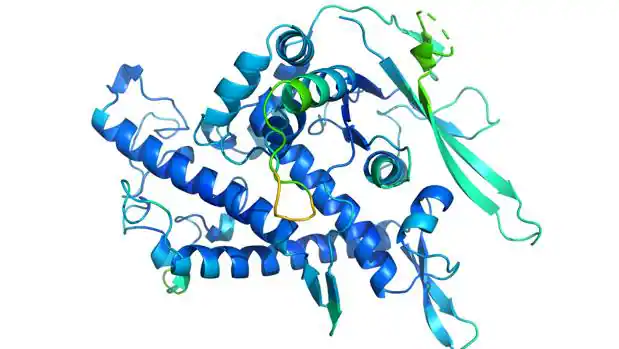
Ajuste entre dos estructuras descubiertas experimentalmente (en verde) y predicha por AlphaFold (en azul)
–
DeepMind
«Hemos estado atascados con este problema —el de cómo se pliegan las proteínas— durante casi 50 años», ha explicado en un comunicado John Moult, presidente y cofundador de CASP, e investigador en la Universidad de Maryland (EE.UU.). «Ver cómo DeepMind ha creado una solución para esto (…) es un momento muy especial».
No se trata de un avance menor. Según ha informado «Nature.com», poder predecir la estructura de una proteína, a partir de su secuencia de aminoácidos sería un grandísimo salto para las ciencias de la vida y la Medicina. Aceleraría enormemente los esfuerzos para entender los ladrillos básicos de la vida haría que la investigación de nuevos medicamentos fuera más rápida y avanzada. Según ha dicho Demis Hassabis, director general de DeepMind: «Creo que ésta es la cosa más significativa que hemos logrado, en términos del impacto que tendrá en el mundo real».
«Es un avance de primer orden, sin duda uno de los resultados científicos más importantes de mi vida», ha comentado para «Nature» Mohammed AlQuraishi, biólogo computacional de la Universidad de Columbia, yu participante en el CASP. Tanto que, al haberse resuelto el problema fundamental, ha dicho que muchos grupos se dedicarán a otra cosa.
Una antigua búsqueda
En 1972, el Nobel en Química Christian Anfinsen postuló que la estructura de una proteína está completamente determinada por su secuencia de aminoácidos. Pero en 1969, Cyrus Levinthal predijo que haría falta más tiempo que la edad del universo para enumerar todas las posibles configuraciones de una proteína típica, por medio de cálculos —predijo que una proteína típica tiene 10^300 conformaciones—. Curiosamente, a pesar de ese número de configuraciones, las proteínas se pliegan a medida que se van produciendo, en los ribosomas, en cuestión de unos pocos milisegundos.
En 1994 se fundó el CASP para acelerar la investigación y poner en común los avances. Desde entonces, cada dos años, en este encuentro se han seleccionado proteínas recientemente determinadas por métodos experimentales, para poner a prueba modelos bioinformáticos predictivos, sin que los desarrolladores pudieran saber cuál era la estructura de la proteína analizada. Para medir el éxito de sus esfuerzos, se desarrolló una medida, conocida como GDT (de Test de Distancia Global, en inglés), cuya puntuación va de cero a 100: aquellas puntuaciones cercanas a 90 suelen ser consideradas como una estimación competente.
En el undécimo cuarto encuentro de CASP la inteligencia artificial AlphaFold ha logrado una puntuación GDT media de 92,4, tras trabajar con varias proteínas. Esta precisión se traduce en que la IA ha cometido un error comparable al ancho de un átomo o 0,1 nanómetros, cuando un nanómetro es la millonésima parte de un milímetro.
Una auténtica revolución
«Estos resultados abren la puerta a que los biólogos usen la predicción computacional de la estructura como herramienta nuclear en la investigación científica», han explicado desde DeepMind. «Nuestros métodos pueden ser especialmente útiles para importantes tipos de proteínas, como las de membrana —son las que se sitúan en la bicapa lipídica que separa el interior de las células del medio exterior— que son especialmente difíciles de cristalizar y, por tanto, de determinar experimentalmente».
«Este trabajo computacional es un avance maravilloso en el problema del plegamiento de las proteínas, un gran reto para la Biología desde hace 50 años»
«Este trabajo computacional es un avance maravilloso en el problema del plegamiento de las proteínas, un gran reto para la Biología desde hace 50 años», ha explicado Venki Ramakrishnan, presidente de la Royal Society. «Y ha ocurrido décadas antes de lo que muchos habían predicho. Será muy emocionante ver las muchas formas como esto cambia fundamentalmente la investigación biológica».
«Lo que el equipo de DeepMind ha logrado hacer es fantástico y cambiará el futuro de la biología estructural y la investigación de proteínas», ha explicado en «Sciencemag.org» Janet Thornton, directora emérita del Instituto Europeo de Bioinformática.
Para lograr estos resultados, el equipo de DeepMind ha estado cuatro años trabajando en crear y entrenar a una red neural capaz de procesar «gráficas espaciales», que representan el plegamiento de las proteínas y las relaciones de sus residuos de aminoácidos. Dicho sistema de IA aprende a refinar estas gráficas empleando secuencias relacionadas, alineamientos de múltiples secuencias y otras representaciones.
Proceso de aprendizaje para procesar gráficas espaciales y predecir la estructura 3D de proteínas
–
DeepMind
Después de múltiples iteraciones, el sistema aprendió a hacer predicciones sobre la estructura física con gran precisión. Para ello, fue necesario entrenarlo con la secuencia de las 170.000 proteínas conocidas y aprovechar también grandes bases de datos de proteínas desconocidas, usando semanas de computación en un número de procesadores comparable a 100 o 200 GPUs o unidades de preocesamiento gráfico. También fue necesario crear «algoritmos de tensión», que permitieron conectar pequeños grupos de aminoácidos, antes de formar un conjunto, como si se tratara de un puzzle resuelto a partir de pequeños grupos.
Ahora, los investigadores de DeepMind están trabajando para publicar sus avances en una revista científica y en descubrir nuevas formas de facilitar el acceso a esta herramienta gran escala. Además, pretenden averiguar cómo estas predicciones pueden contribuir a estudiar ciertas enfermedades, a facilitar el desarrollo de medicamentos y complementar métodos experimentales ya existentes.
No obstante, AlphaFold todavía no es perfecto. Por ejemplo, tiene problemas con estructuras formadas por la repetición de pequeños segmentos, y todavía no permite estudiar complejos de varias proteínas que desempeñan juntas su función en las células.
Un futuro prometedor
Esta IA no es solo un potente «telescopio» con el que adentrarse en el desconocido universo de millones de proteínas cuya estructura no se ha revelado. Con la vista puesta en el futuro, los investigadores de DeepMind han comentado que esta herramienta puede ser útil para responder a futuras pandemias, puesto que AlphaFold pudo predecir con éxito las estructuras de ORF8 y ORF3a, dos proteínas del SARS-CoV-2.
Todavía más allá, han sugerido que la IA podrá ser de ayuda para estudiar cómo las proteínas interaccionan con el ADN, el ARN u otras moléculas.
«Los sistemas como AlphaFold demuestran el increíble potencial de las IAs como herramienta para permitir descubrimientos fundamentales», han concluido. «(…) Hay muchos aspectos de nuestro universo que son desconocidos. El avance anunciado ahora nos da más seguridad de que la IA se convertirá en una de las herramientas más útiles para expandir las fronteras del conocimiento científico».